


A series of substituted ferrocenyl boron derivatives was synthesized. The oxidation of the ferrocenyl unit resulted in a significant increase of the boron-centered Lewis acidity. The neutral and cationic Lewis acids were characterized by NMR-spectroscopy, crystal structure analysis and by computational methods. The new Lewis acids were then applied in the Meinwald rearrangement of epoxides, predominantly furnishing aldehydes as the kinetic products.
We conducted an investigation into the palladium-catalyzed carbon-sulfur cross-coupling reaction involving a 2-bromothiophene derivative and potassium thioacetate as a substitute for hydrogen sulfide. This investigation utilized kinetic and computational methods. We synthesized two palladium complexes supported by the bisphosphane ligands bis(diphenylphosphino)ferrocene (DPPF) and bis(diisopropylphosphino)ferrocene (DiPPF), as well as their tentative intermediates in the catalytic cycle. Reaction rates were measured and then compared to computational predictions.
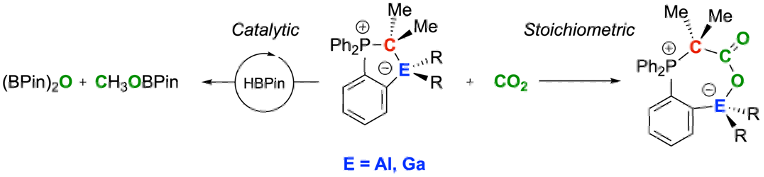
We report on so-called "hidden FLPs" (FLP: frustrated Lewis pair) consisting of a phosphorus ylide featuring a group 13 fragment in the ortho position of a phenyl ring scaffold to form five-membered ring structures. Most of the title compounds readily react with carbon dioxide to provide stable adducts, although the formation of the Lewis acid/base adducts was observed in the solid state. Strikingly, 0.3–3.0 mol% of the reported aluminum and gallium/carbon-based ambiphiles catalyze the reduction of CO2 to methanol with satisfactory high selectivity and yields using pinacol borane as stoichiometric reduction equivalent. Comprehensive computational studies provided valuable mechanistic insights and shed more light on activity differences.

Despite recent achievements in the field of frustrated Lewis pairs (FLPs) for small molecule activations, the reversible activation and catalytic transformations of N–H-activated ammonia remain a challenge. Here we report on a rare combination of an aluminium Lewis acid and a carbon Lewis base. A so-called hidden FLP consisting of a phosphorus ylide featuring an aluminium fragment in the ortho position of a phenyl ring scaffold is introduced. Although the formation of the Lewis acid/base adduct is observed in the solid state, which at first glance leads to formally quenched FLP reactivity, we show that the title compound readily reacts with non-aqueous ammonia thermoneutrally and splits the N–H bond reversibly at ambient temperature. In addition, NH3 transfer reactions mediated by a main-group catalyst are presented. This proof-of-principle study is expected to initiate further activities in utilizing N–H-activated ammonia as a readily available, atom-economical nitrogen source.
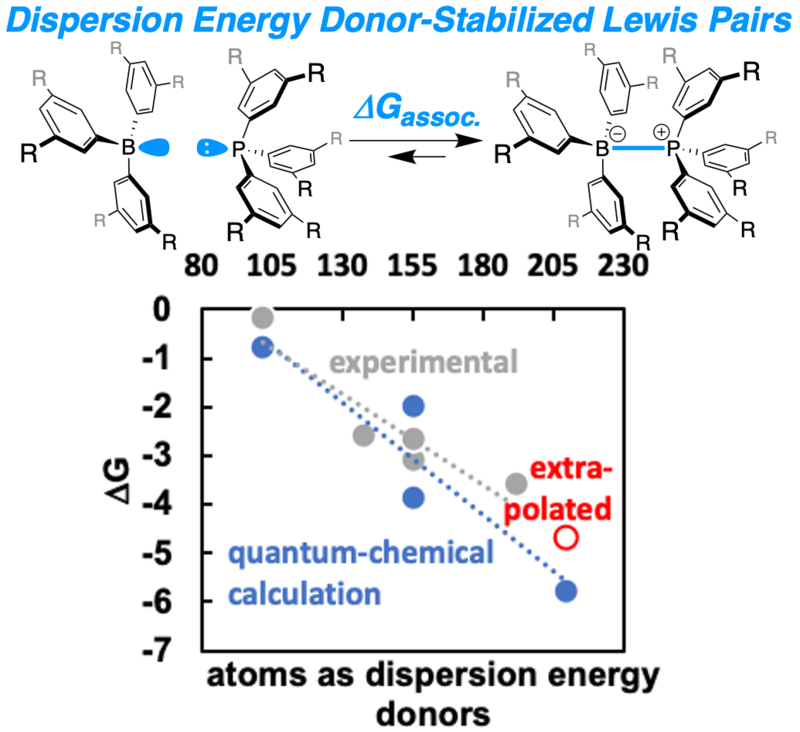
An isostructural series of boron/phosphorus Lewis pairs was systematically investigated. The association constants of the Lewis pairs were determined at variable temperatures, enabling the extraction of thermodynamic parameters. The stabilization of the Lewis adduct increased with increasing size of the dispersion energy donor groups, although the donor and acceptor properties of the Lewis pairs remained largely unchanged. This data was utilized to challenge state-of-the-art quantum chemical methods, which finally led to an enhanced workflow for the determination of thermochemical properties of weakly bound Lewis pairs within an accuracy of 0.6 to 1.0 kcal•mol–1 for computed association free energies.
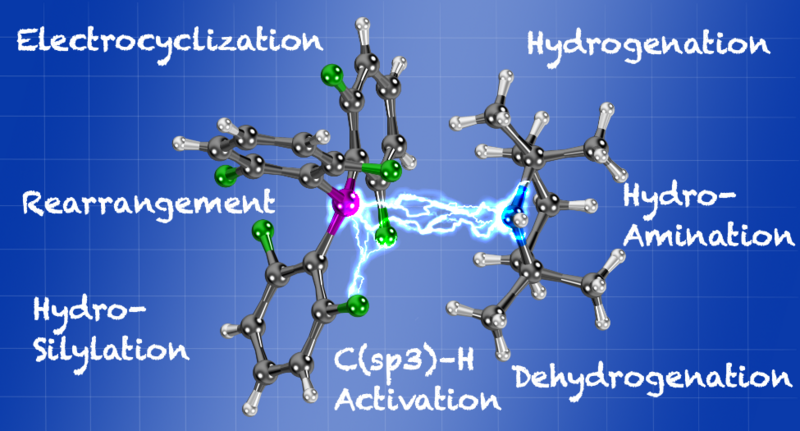
In this Account, we provide detailed insight into how Lewis acidity and Lewis basicity correlate to reactivity. The reactivity of frustrated Lewis pairs will be systematically discussed in context with selected reactions. The influence of major electronic modifications of the Lewis pairs is correlated with the ability to activate molecular hydrogen, to channel reaction kinetics and reaction pathways or to achieve C(sp3)–H activations.

A series of redox-responsive ferrocenyl-substituted boranes and boronic esters were synthesized. Oxidation of the ferrocenyl ligand to the ferrocenium resulted in drastic increase of the Lewis acidity beyond the strength of SbF5, which was investigated experimentally and computationally. The resulting highly Lewis acidic boron compounds were used for catalytic C–F and S–F bond activation.
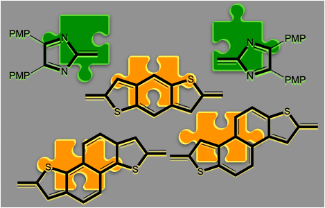
The synthesis of three novel imidazolyl-substituted sulfurcontaining heteroacenes is reported. These heteroacenes consisting of annelated benzo- and naphthothiophenes serve as precursors for the generation of open-shell quinoid heteroacenes by oxidation with alkaline ferric cyanide. Spectroscopic and computational experiments support the formation of reactive open-shell quinoids, which, however, quickly produce paramagnetic polymeric material.
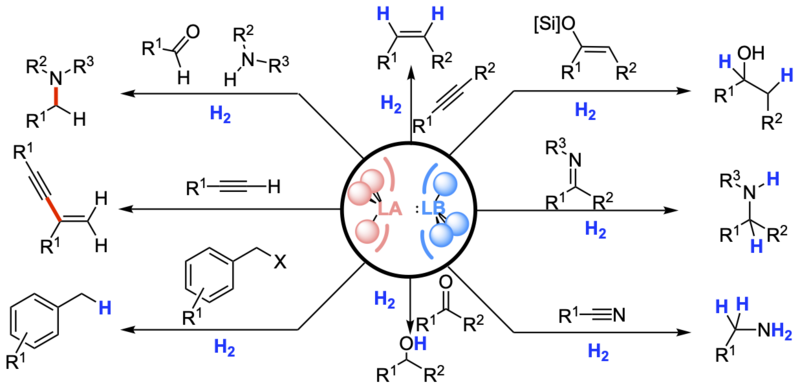
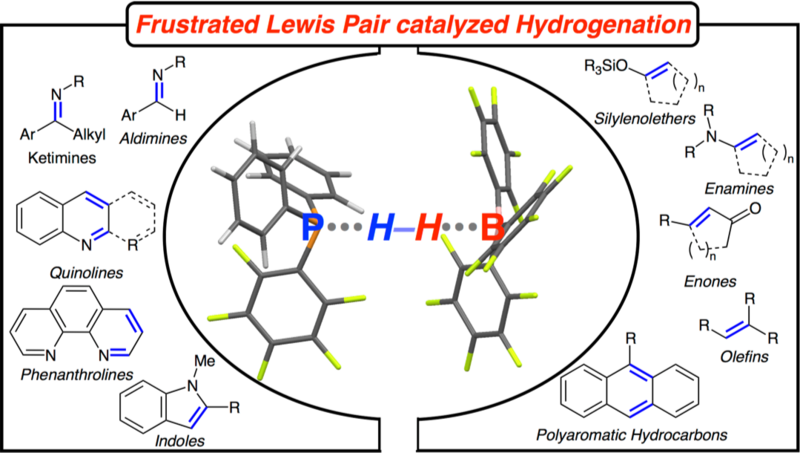
In recent years, frustrated Lewis pairs have been widely used in small molecules activation and catalytic transformations. This graphic review is aimed to provide the fundamental understanding of frustrated Lewis pair reactivity and the exploitation thereof in catalytic reactions.
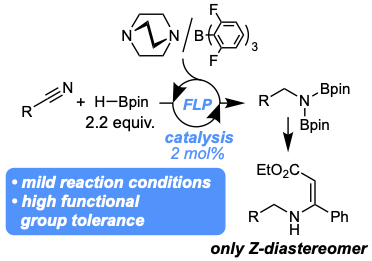
A frustrated Lewis pair-catalyzed hydroboration of aromatic and aliphatic nitriles was developed. The catalyst provides the primary amines in high yields of 77–99% with catalyst loading as low as 2 mol%. The reaction displays high functional group tolerance towards esters, amides, nitro groups and aliphatic halides. The addition of the diborylated amines to ethyl 3-phenylpropiolate proceeds with Z-selectivity with d.r. of>99:1 in 77–90% yield over two steps. The reaction mechanism was investigated by control and computational experiments.

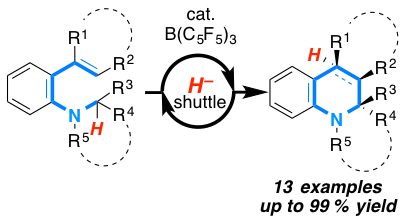
The stereospecific sigmatropic [1,5] carbon shift of C3 ammonium enolates is discovered. According to mechanistic, kinetic and computational experiments, this new rearrangement proceeds via the catalytic generation of a transient C3 ammonium enolate by intramolecular aza-Michael addition. This intermediate rapidly undergoes [1,5] sigmatropic carbon migration to furnish the respective tetrahydroquinoline-4-ones with excellent diastereoselectivities of d.r. > 99:1 and in 61-98% yield.

The synthesis and characterization of a homologous series of quinoid sulfur-containing imidazolyl-substituted heteroacenes is described. The optoelectronic and magnetic properties were investigated by UV/vis, fluorescence and EPR spectroscopy as well as quantum chemical calculations and were compared to the corresponding benzo congener. The room-temperature and atmosphere-stable quinoids display strong absorption in the NIR region between 678–819 nm. The dithieno[3,2- b :2',3'- d ]thiophene derivative and the thieno[2',3':4,5]thieno[3,2- b ]thieno[2,3- d ]thiophene derivative were EPR active at room temperature. For the latter, variable temperature EPR spectroscopy revealed the presence of a thermally accessible triplet state, with a singlettriplet separation of 14.1 kJ/mol.
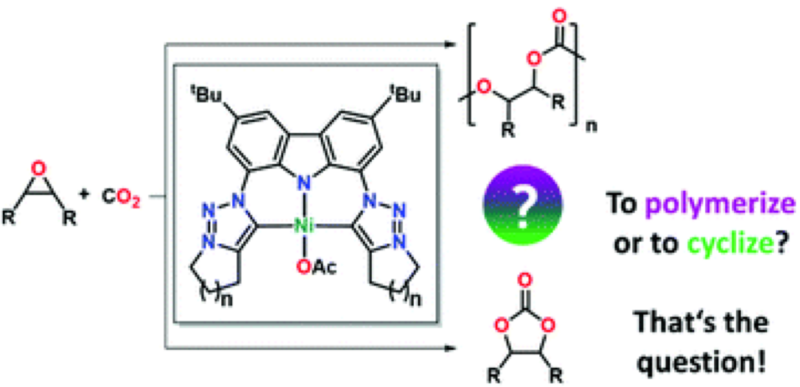
We report the syntheses of two rigid mesoionic carbene (MIC) ligands with a carbazole backbone via an intramolecular Finkelstein-Cyclisation cascade and investigate their coordination behavior towards nickel(II) acetate. Despite the nickel(II) carbene complexes 4a,b show only minor differences in their chemical composition, they display curious differences in their chemical poperties, e.g. solubility. Furthermore, the potential of these novel MIC complexes in the coupling of carbon dioxide and epoxides as well as the differences in reactivity compared to classical NHC-derived complexes are evaluated.
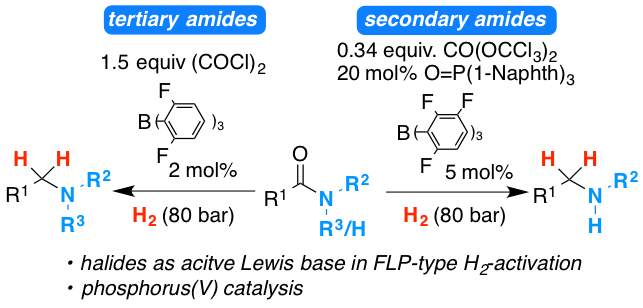
The development of the frustrated Lewis pair catalyzed hydrogenation of tertiary and secondary amides is reviewed. Detailed insight into our strategies in order to overcome challenges during the reaction development process is provided. Furthermore, the developed chemistry is extended to the hydrogenation of poly amides and trifluoroacetyl amides for the convenient introduction of trifluoro ethyl groups into organic molecules.
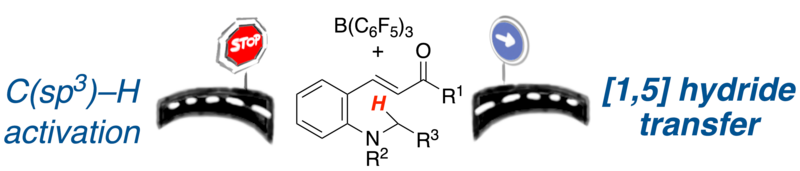
The borane catalyzed redox isomerization of 2-amino chalcones is developed. The tetrahydroquinolines are obtained in high yield as a mixture of cis/trans diastereomers in a ratio of 2:1 to >95:5. The reaction mechanism is investigated by mechanistic, kinetic and computational methods, concluding that the reaction proceeds through concerted [1,5] hydride shift in contrast to borane-mediated C(sp3)–H hydride abstraction.
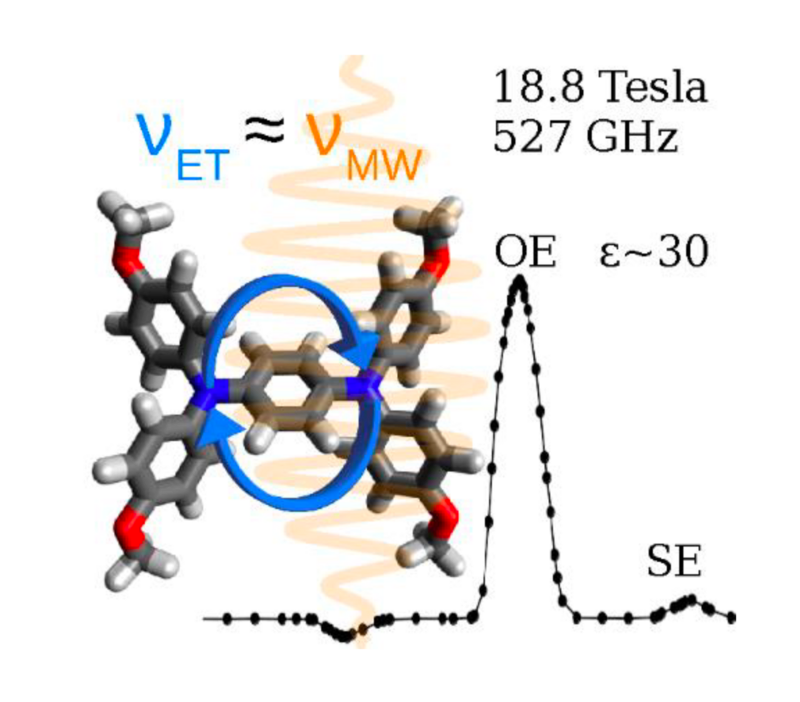
Mixed-valence compounds were investigated as novel polarizing agents for DNP NMR. These compounds constitute a group of molecules in which molecular mobility persists even in solids. Consequently, they can be used to perform Overhauser-DNP NMR experiments in solid state, and they specifically show favorable properties for applications at ultra-high magnetic fields (DNP=dynamic nuclear polarization, NMR=nuclear magnetic resonance).
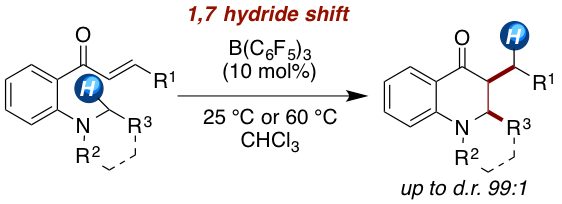
[51] "Diastereoselective Synthesis of Dihydro-quinolin-4-ones by a Borane-Catalyzed Redox-Neutral endo-1,7-Hydride Shift" Garrit Wicker, Roland Schoch, Jan Paradies Org. Lett. 2021, 23 3626–3630. DOI:10.1021/acs.orglett.1c01018.
The borane-catalyzed synthesis of dihydroquinoline-4-ones is developed. The amino-substituted chalcones undergo a 1,7-hydride shift upon Lewis acid activation to form a zwitterionic iminium enolate, which collapses to the dihydroquinoline-4-one scaffold. The reaction proceeds in high yields (75–99%) with an excellent diastereoselectivity of up to >99:1 (cis:trans). The reaction mechanism is investigated by kinetic, isotope labeling, and computational experiments.
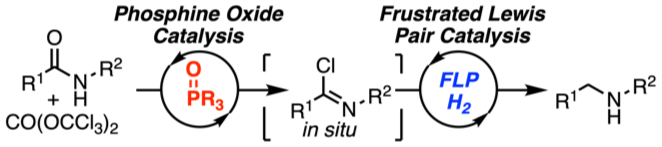
The metal‐free catalytic hydrogenation of secondary carboxylic acid amides is developed. The reduction is realized by two new catalytic reactions. First, the amide is converted into the imidoyl chloride by triphosgene (CO(OCCl3)2) using novel phosphorus(V) catalysts. Second, the in situ generated imidoyl chlorides are hydrogenated in high yields by an FLP‐catalyst. Mechanistic and quantum mechanical calculations support an autoinduced catalytic cycle for the hydrogenation with chloride acting as unusual Lewis base for FLP‐mediated H2‐activation.
The potential of two chiral amidines and three non-chiral boranes in the metal-free hydrogen activation was explored. The resulting chiral amidiunium borohydride salts were investigated in asymmetric hydrogenation reactions of ketimines, activated double bonds and dehydro amioacid esters.
Lysophosphatidic acid (LPA) as the biomarker of early stage ovarian cancer is essentially difficult to be detected due to lack of target spots. A dually crosslinked supramolecular hydrogel (DCSH) was developed to achieve sensing of LPA, which act as a competitive guest molecule triggering the responsive crosslinking of the DCSH. Through this strategy, the surface plasmon resonance combined with optical waveguide spectroscopy could be used to quantitatively detect LPA with a responsive range covering physiological conditions (in pure form as well as mimicking LPA plasma solution) with high selectivity and sensitivity. LPA efficiently insert into the host molecule β-cyclodextrin (β-CD) up to a 1:2 ratio as the competitive interaction mechanism confirmed by onedimensional nuclear overhauser effect spectroscopy (1D NOESY), high resolution mass spectrometry (HRMS), isothermal titration calorimetry (ITC) and computational simulation. Our method opens a new strategy to detect biomarkers without target spot and provides a platform for surface plasmon resonance (SPR) based sensors measuring small molecules.

The palladium catalyzed C–S cross coupling reaction is investigated as novel efficient tool for the synthesis of poly(phenyl)sulfide derivatives. The reaction proceeds through the polycondensation of dibromo arenes with a H2S surrogate to yield poly(aryl)sulfides. The reaction is generalized by the synthesis of so far unprecedented poly(2,5-thiophene)sulfide. Number average molecular weights of up to 3780 and 1770 g mol−1 for poly(phenyl)sulfide and poly(thiophene)sulfide are achieved with degrees of polymerization (DPn) of 10 and 7, respectively. A mechanism for the new polycondensation reaction is suggested.

Very high activities were observed in the redox-induced hydroamination of alkynes by employing a redox-active gold(I) complex featuring an electron-deficient, terphenyl-substituted phosphonitebased ligand. The hydroamination proceeds roughly two-fold faster with the in situ oxidized catalysts than with their reduced form.

The coexistence of a strong Lewis acid and a Lewis base in solution, the so called frustrated Lewis pair, has led to the discovery of metal-free hydrogen activation. Since then, this observation has inspired numerous chemists to develop more examples. Metal-free hydrogenation is so far the most studied application of frustrated Lewis pairs in chemistry and highly efficient methodologies for a number of substrates have been developed. However, the targeted choice of a FLP-catalyst is yet rather intricate, due to the lack of an in depth understanding of FLP-reactivity. The presented structure-reactivity-relationship for hydrogenation reactions allowed the targeted development and optimization of unprecedented reactions using FLPs as catalysts. This article provides insight into FLP-reactivity by summarizing our mechanistic and synthetic work in this field.
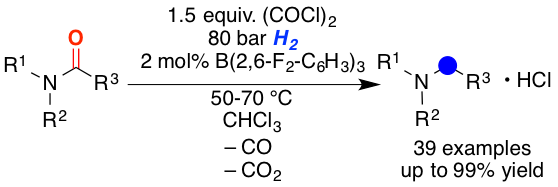
A method for the metal-free reduction of carboxylic amides using oxalyl chloride as activating agent and hydrogen as final reductant is introduced. The reaction proceeds via the hydrogen splitting by B(2,6-F2-C6H3)3 in combination with chloride as the Lewis base. Density functional theory calculations support the unprecedented role of halides as active Lewis base components in the frustrated Lewis pair mediated hydrogen activation. The reaction displays broad substrate scope for tertiary benzoic acid amides and alpha-branched carboxamides.
The borane-catalyzed synthesis of quinoline derivatives bearing tetrasubstituted stereocenters from vinyl anilines has been developed. Mechanistic studies and quantum-mechanical investi¬gations support the hydride abstraction/electro¬cycli¬zation/hydride addition mechanism. The products were obtained in up to 99% yield with a diastereoselectivity of >99% in favour for the 3a-5-cis isomer.
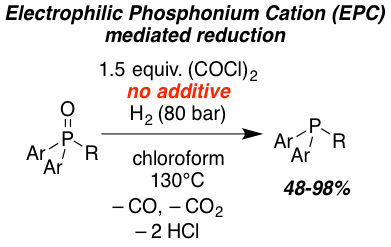
The metal-free reduction of phosphane oxides with molecular hydrogen (H2) using oxalyl chloride as activating agent was achieved. Quantum-mechanical investigations support the heterolytic splitting of H2 by the in situ formed electrophilic phosphonium cation (EPC) and phosphane oxide and subsequent barrierless conversion to the phosphane and HCl. The reaction can also be catalyzed by the frustrated Lewis pair (FLP) consisting of B(2,6-F2C6H3)3 and 2,6-lutidine or phosphane oxide as Lewis base. This novel reduction was demonstrated for triaryl and diaryl phosphane oxides providing access to phosphanes in good to excellent yields (51-93%).
This review summarizes the principle reaction mechanisms of frustrated Lewis pairs (FLP) in hydrogenations and carbon-nitrogen and carbon-carbon bond forming reactions. The fundamental mechanism of hydrogen activation by FLPs is reviewed and the influence of the FLP’s nature on hydrogenation reactions is discussed, leading to a structure-reactivity relationship in phosphine/borane or amine/borane derived FLPs. This reactivity concept is validated for a series of FLP-catalyzed reactions. Furthermore, alternative reaction mechanisms e.g. protodeborations or sigma-bond metathesis are discussed.
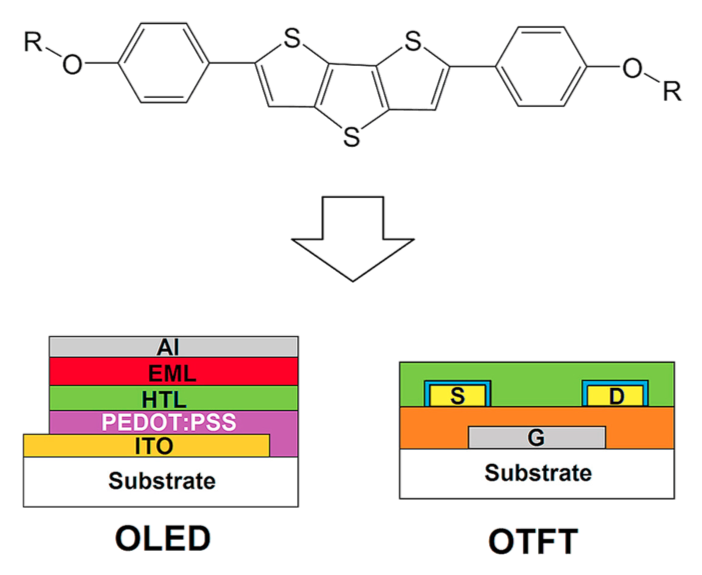
The synthesis and mesophase characterization of two 2,6-bis(4-(alkyloxy)phenyl)dithieno [3,2-b:2′,2′-d]thiophenes indicate successful application of a palladium-catalyzed carbon-sulfur cross-coupling/5-endo-dig cyclization sequence and the appearance of a nematic mesophase in the resulting products. Spectroscopic and cyclovoltammetric measurements reveal molecular orbital energies that are comparable to a good standard hole conductor. Consequently, one of the compounds was successfully applied as a hole conducting layer in an electroluminescent sample. In thin-film transistors (TFT), the two compounds show charge carrier mobilities in the saturation region μsat of 0.13 cm2V−1s−1 and 0.03 cm2V−1s−1, respectively. The injection barrier at the metal semiconductor interface could be reduced by alkanethiol treatment, thereby enabling a linear ID (VD) characteristics in the low VD region and maximum currents up to ID,max = - 18 μA at VG = - 2.5 V in the saturation regime. Inverter circuits in single-type TFT technology were implemented.
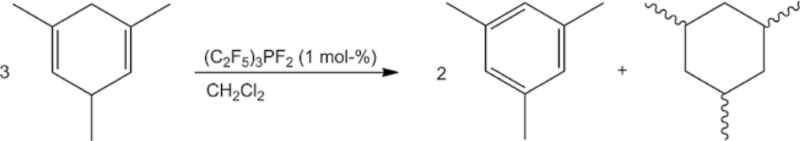
Transfer hydrogenation plays an important part in organic chemistry. Recently, strong Lewis acids like B(C6F5)3 have been introduced as a catalyst for these reactions. We successfully employed the Lewis acid (C2F5)3PF2 as a catalyst in the transfer hydrogenation between 1,3,5‐trimethylcyclohexa‐1,4‐diene and 1,1‐diphenylethylene. Surprisingly, the treatment of the diene alone with a catalytic amount of (C2F5)3PF2 led to a quantitative dismutation to mesitylene and 1,3,5‐trimethylcyclohexane. With B(C6F5)3, there was a solvent‐dependency: in CH2Cl2 mainly the dismutation products were obtained, while in toluene the evolution of H2 was observed. Additionally, the catalytic activity of various perfluoroalkylated germanes and silanes was tested.
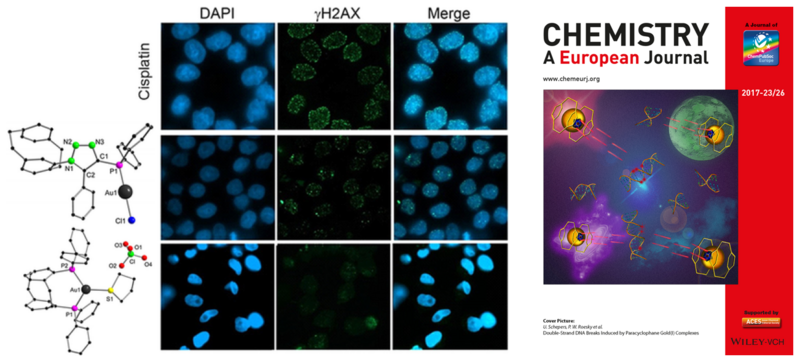
Gold(I) complexes of ClickPhos [2.2]paracyclophane ligands were synthesized in excellent yields and fully characterized by spectroscopic methods as well as X‐ray crystallography. The complexes exhibit a rigid ligand backbone and a triazolyl moiety and were systematically studied with respect to their cytotoxic properties. In combination with the ionic complex [(GemPhos)Au(tht)][ClO4] (tht=tetrahydrothiophene), in which the gold(I) atom exhibits a distorted trigonal coordination sphere of two phosphines and a labile tht ligand, their efficiency in cytotoxicity was investigated in HeLa, MCF7, and HCT116 cells as well as in a zebrafish model. Their cytotoxicity and their mechanisms of action are different and involve apoptosis, necrosis, and DNA damage. The compounds presented herein are potent metal‐based cytostatics displaying LD50 values from 3.5–38 μm in different tumor cell lines and induce double‐strand DNA breaks (DSB) as shown by H2AX phosphorylation (γH2AX) at foci of DSBs.
[37] "Borane-catalyzed indole synthesis through intramolecular hydroamination" S. Tussing, M. Ohland, G. Wicker, U. Flörke, J. Paradies* Dalton Trans. 2017, 46, 1539-1545.
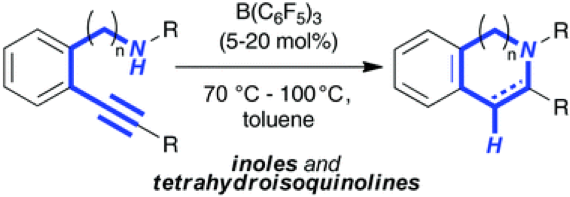
The reaction of 2-alkynyl anilines with catalytic amounts of B(C6F5)3 (5 mol%) resulted in the formation of 2-substituted indoles according to an intramolecular hydroamination in good to excellent yields. Reaction intermediates as well as products were characterized by NMR spectroscopy and by X-ray crystallography. The domino hydroamination/hydrogenation sequence allowed the efficient synthesis of tetrahydroquinoline in good yield.
[36] "Heteroacene synthesis through C–S cross coupling / 5-endo-dig cyclization" Peter Oechsle, Ulrich Flörke, Hans Egold, and Jan Paradies Chem. Eur. J. 2016, 51, 18559-18563.
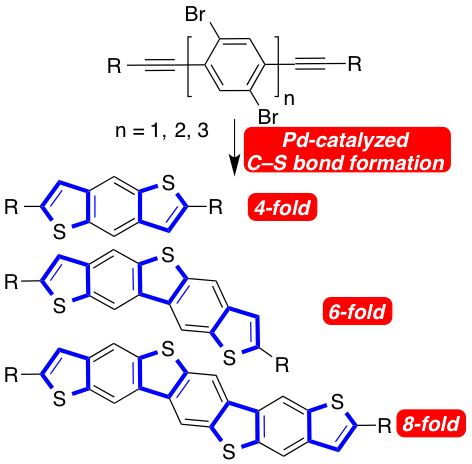
The highly efficient, one step of sulfur-containing heteroacenes was achieved through palladium-catalyzed C–S cross coupling of bisalkynes with thioacetate as hydrogen sulfide surrogate. The heteroacenes consisting of three, five and seven fused aromatic rings were obtained in one single catalytic step by four-, six- and eight-fold C–S bond formation.
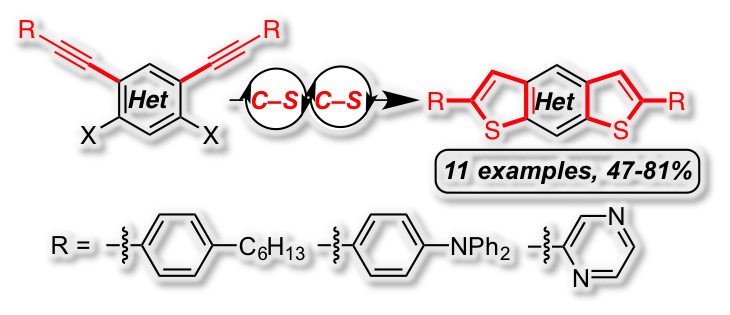
[35] “Concise synthesis of dithiophene derivatives by palladium-catalyzed multiple C–S cross coupling/cyclization sequence” Peter Oechsle,a Peng Hou,a Ulrich Flörkeb and Jan Paradiesa*, Adv. Synth Catal. 2016, 7, 3770-3776, DOI:10.1002/adsc.201600802
The facile synthesis of new sulfur-containing fused heterocycles was achieved by a twofold domino reaction consisting of a carbon-sulfur cross coupling, followed by a 5-endo-dig cyclization. Using this strategy a series of benzo, thiopheno, pyridino and pyrazino dithienoacenes with electron-neutral (-C6H4-nHex), electron-rich (-C6H4-NPh2) and electron-deficient (-C4H3N2) substituents were synthesized in high yields. The developed method was applied in the efficient synthesis of a complex donor-acceptor molecule. The photophysical and electrochemical properties of the products were analyzed by UV-VIS/luminescence spectroscopy and cyclic voltammetry.
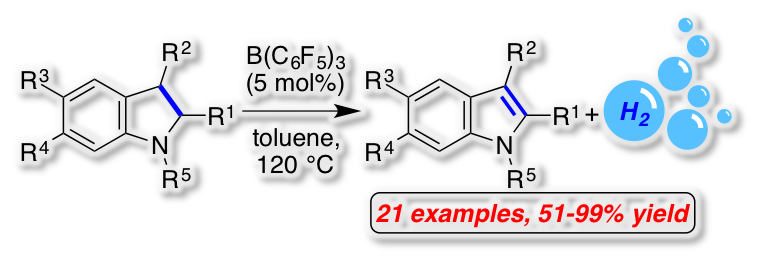
[34] "Frustrated Lewis Pair-catalyzed dehydrogenative oxidation of indolines and other heterocycles" Alexander F. G. Maier, Sebastian Tussing, Tobias Schneider, Ulrich Flörke, Zheng-Wang Qu, Stefan Grimme* and Jan Paradies* Angew. Chem. 2016, 55, 12219–12223. The FLP-catalyzed acceptorless dehydrogenation of heterocycles was developed. The oxidation with concomitant liberation of molecular hydrogen proceeded in high to excellent yields for N-protected indolines as well as four other substrate classes. The mechanism of this unprecedented FLP-catalyzed reaction was investigated by mechanistic studies, characterization of reaction intermediates by NMR spectroscopy and x-ray crystal analysis and by quantum-mechanical calculations. The hydrogen liberation from the intermediary formed ammonium hydroborate is the rate determining step of the oxidation. The addition of a weaker Lewis-acid as hydride shuttle increased the reaction rate by 2.28 though a second catalytic cycle.
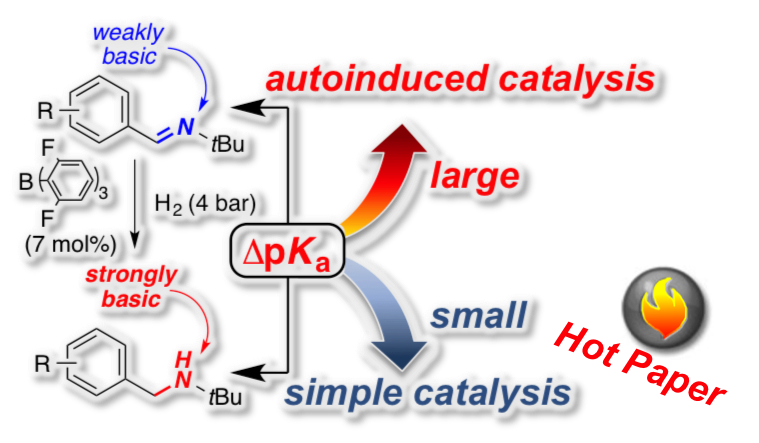
[33] "Structure-reactivity relationship in the FLP-catalyzed hydrogenation of imines" Sebastian Tussing, Karl Kaupmees and Jan Paradies* Chem. Eur. J. 2016, 22, 7422–7426
The autoinduced, frustrated Lewis pair (FLP)-catalyzed hydrogenation of 16 benzene ring substituted N-benzylidene-tertbutylamines with B(2,6-F2C6H3)3 and molecular hydrogen was investigated by kinetic analysis. The pKa values for imines and for the corresponding amines were determined by quantum-mechanical methods and provided a direct proportional relationship. The correlation of the two rate constants k1 (simple catalytic cycle) and k2 (autoinduced catalytic cycle) with pKa difference between imine and amine pairs (ΔpKa) or Hammett’s σ parameter served as useful parameters to establish a structure-reactivity relationship for the FLP-catalyzed hydrogenation of imines.
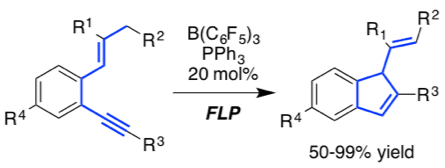
[32] "Frustrated Lewis Pair-catalyzed cycloisomerization of 1,5-enynes via 5-endo-dig cyclization/protodeborylation sequence" Sergej Tamke, Zheng-Wang Qu, Nikolai A. Sitte, Ulrich Flörke, Stefan Grimme,* and Jan Paradies* Angew. Chem. 2016, 55, 4335-4339
The first frustrated Lewis pair-catalyzed cycloisomerization of a series of 1,5-enynes was developed. The reaction proceeds via the p-activation of the alkyne and subsequent 5-endo-dig cyclization with the adjacent alkene. The presence of PPh3 was of utmost importance on the one hand to prevent side reactions (e.g. 1,1-carboboration) and on the other hand for the efficient protodeborylation to achieve the catalytic turnover. The mechanism is explained on the basis of quantum-chemical calculations which are in full agreement with the experimental observations.
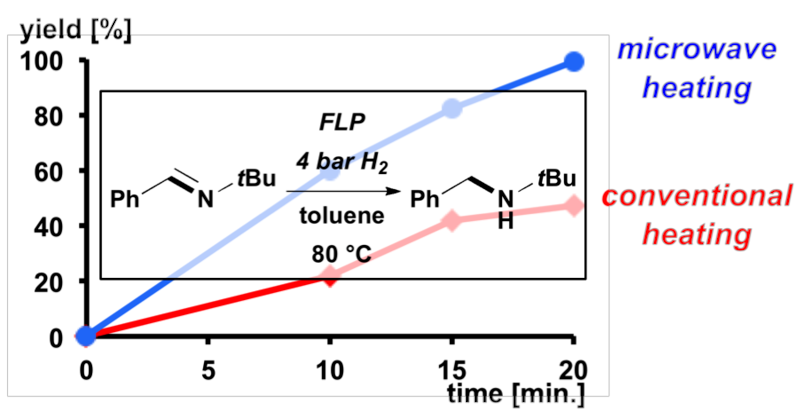
FLP-catalyzed hydrogenations of 15 substrates were compared using microwave irradiation and conventional heating. The direct comparision revealed that a rate acceleration of up to 2.5 was achieved in the presence of microwaves. This heating method is particulary promising for the hydrogenation of nitrogen-containing heterocycles. Acridine, quinines and especially 1-methyl indole were reduced very efficiently under mild conditions and only 4 bar hydrogen pressure in high yields.
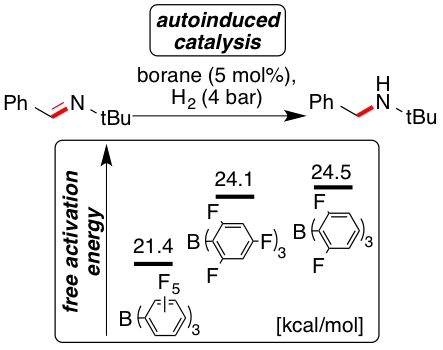
The frustrated Lewis pair (FLP)-catalyzed hydrogenation and deuteration of N-benzylidene-tert-butylamine (2) was kinetically investigated by using the three boranes B(C6F5)3 (1), B(2,4,6-F3-C6H2)3 (4), and B(2,6-F2-C6H3)3 (5) and the free activation energies for the H2 activation by FLP were determined. Reactions catalyzed by the weaker Lewis acids 4 and 5 displayed autoinductive catalysis arising from a higher free activation energy (2 kcal mol−1) for the H2 activation by the imine compared to the amine. Surprisingly, the imine reduction using D2 proceeded with higher rates. This phenomenon is unprecedented for FLP and resulted from a primary inverse equilibrium isotope effect.
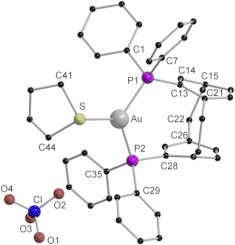
GemPhos, a diphosphine ligand with a rigid paracyclophane scaffold, was used to prepare complexes of the noble metals gold, palladium, and platinum. The reaction of GemPhos with [Au(tht)2][ClO4] (tht = tetrahydrothiophene) surprisingly yielded a mononuclear charge separated gold compound [(GemPhos)Au][ClO4], in which the gold atom exhibits an uncommon trigonal planar coordination geometry. Furthermore, similar palladium [(GemPhos) (PdCl2)] and platinum [(GemPhos)(PtCl2)] complexes were obtained in very good yields by the reaction of GemPhos with [MCl2(COD)] (M = Pd, Pt; COD = 1,5-cyclooctadiene) in hot DMSO. All compounds were fully characterized by analytical and spectroscopic techniques and their solid-state structures were established by single X-ray crystallography. Their photoluminescent properties were measured at low and ambient temperatures, revealing different behavior depending on the metal and coordination mode.
[28] "Frustrated Lewis Pair catalyzed Hydrosilylation and Hydrosilane mediated Hydrogenation of Fulvenes"S. Tamke, G.-C. Daniliuc, J. Paradies* Org. Biomol. Chem. 2014, 12, 9139-9144.
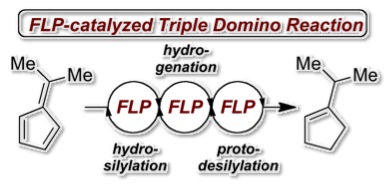
The frustrated Lewis pair (FLP) mediated hydrosilylation of pentafulvenes is described yielding allyl silanes with high regioselectivity in excellent yields. While phenyl substituted allyl silanes undergo B(C6F5)3-mediated rearrangement to vinyl silanes dimethyl derivatives experience FLP-catalyzed hydrogenantion followed by an unprecedented protodesilylation. This observation allowed the metal-free hydrogenation of 6,6-dimethylfulvene to iso-propyl cyclopentene according to a FLP-catalyzed triple domino reaction consisting of hydrosilylation, hydrogenation and protodesilylation. The mechanisms were investigated by deuteration experiments.
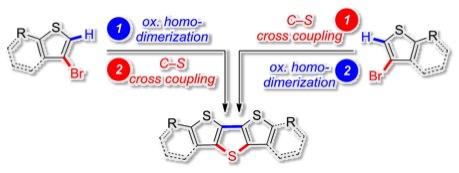
[27] “Ambidextrous Catalytic Access to Dithieno[3,2‑b:2′,3′‑d]thiophene (DTT) Derivatives by Both Palladium-Catalyzed C−S and Oxidative Dehydro C−H Coupling" Peter Oechsle, Jan Paradies* Org. Lett. 2014, 16, 4086–4089.
A modular two-step synthesis of dithieno[3,2-b:2′,3′-d]thiophene (DTT) derivatives by C−S cross-coupling and oxidative dehydro C−H coupling is herein described. Dibenzo[d,d′]thieno[3,2-b;4,5-b′]dithiophene (DBTDT) and associated two donor (anisyl) and acceptor (acetyl) substituted DTT derivatives were synthesized by palladium-catalyzed cross-coupling sequences in 17% to 71% yield over two steps. The 5,5′-disubstituted DTT derivatives were characterized in terms of their photophysical (UV and fluorescence spectroscopy) and electrophysical (cyclovoltammography) properties.
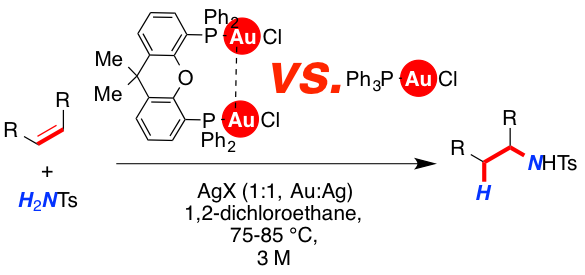
Mono- and dinuclear gold catalysts were investigated in the intermolecular hydroamidation of olefins. Upon activation of [Ph3PAuCl] and [xantphos(AuCl)2] with various silver salts (AgOTf, Ag[BF4] and Ag[SbF6]), diverging reactivity of the resulting cationic gold-complexes was observed. It was found that both the binding ability of the counterion and the solvent have significant impact on the reactivity of the mono- and dinuclear complexes.
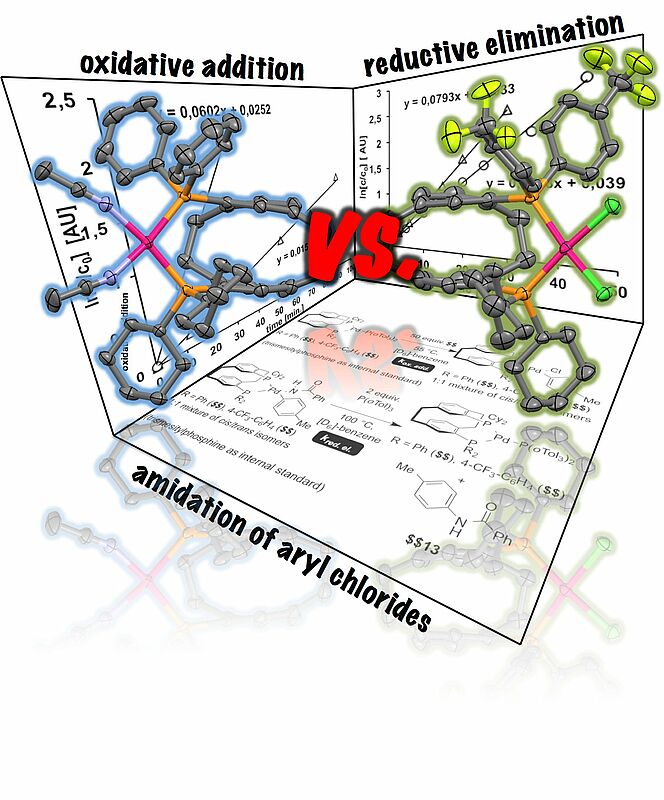
The rate-determining step of the palladium-catalyzed amidation of 4-tolyl chloride with benzamide in the presence of unsymmetrical bisphosphines was investigated. Isostructural [2.2]¬para¬cyclophane-derived bisphosphines bearing dicyclo¬hexyl¬phos¬phino and diarylphosphino moieties were investigated as ligands in the oxidative addition and reductive elimination by kinetic studies. The reductive elimination was accelerated when a bisphosphine was applied as ligand, which contained an electron-rich and a less electron-releasing phosphino-moiety. Apart from the reductive elimination the transmetallation had most severe impact on the palladium-catalyzed amidation of aryl halides.
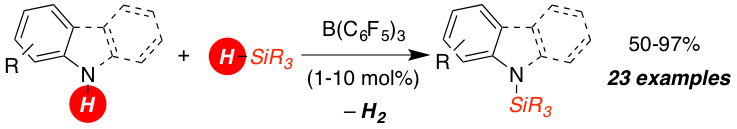
The metal-free B(C6F5)3 catalyzed dehydrocoupling of hydrosilanes with anilines, carbazoles and indoles is reported. For anilines and carbazoles the reaction proceeds by the liberation of H2 as sole Si–N coupling byproduct. Indoles react with diphenyl(methyl) hydrosilane to N-silyl indolines with high diastereoselectively (d.r. 10:1) in excellent yields. A mechanism of this Si–N coupling/hydrogenation sequence is proposed.
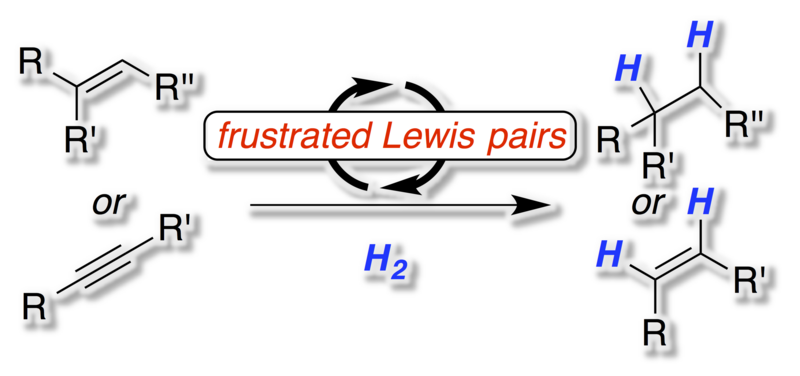
[23] "Metal-free Hydrogenation of Unsaturated Hydrocarbons Employing Molecular Hydrogen" J. Paradies Angew. Chem. 2014, 126, 3624-3629; Angew. Chem. Int. Ed. 2014, 53, 3552-3557.
The metal-free activation of hydrogen by frustrated Lewis pairs (FLPs) is a valuable method for the hydrogenation of polarized unsaturated molecules ranging from imines, enamines, silyl enol ethers to heterocycles. However, one of the most important applications of hydrogenation technology is the conversion of unsaturated hydrocarbons to alkanes or alkenes. Despite the fast development of the FLP-chemistry such reactions proved as highly demanding. This Minireview will provide an overview of the basic concepts of FLP-chemistry, the challenge in the hydrogenation of unsaturated hydrocarbons and first solutions to this central transformation.
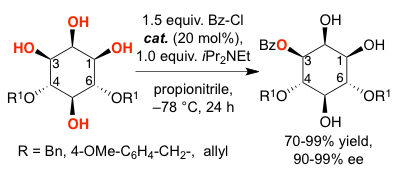
The asymmetric desymmetrization of 4,6-diprotected myoinositol derivatives was achieved by a bifunctional, readily available nucleophilic catalyst. The orthogonally protected products were obtained in 80-99% yield with 90-99% ee. Such structures serve as potential enantiopure building blocks for the synthesis of myo-inositol phosphates.
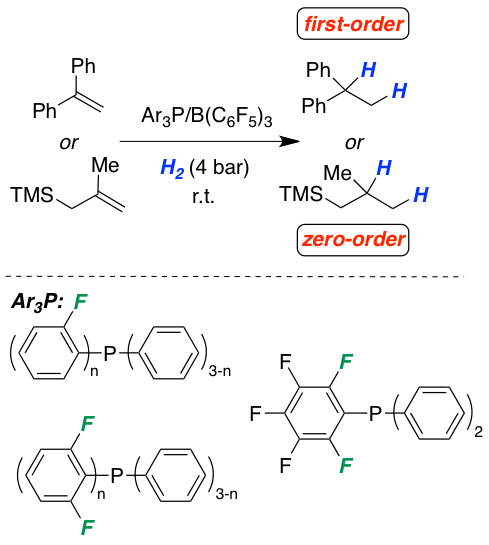
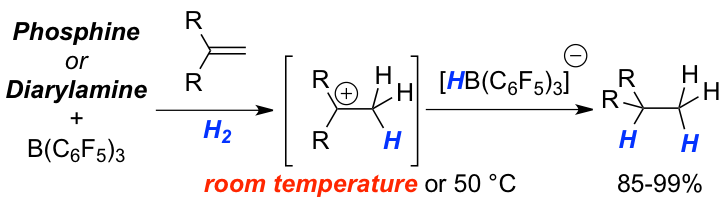
The frustrated Lewis pair-mediated reversible hydrogen activation is studied as a function of the electron-donor quality of a series of phosphines. The increasing acidity of the generated phosphonium species leads to a stepwise lowering of the temperature for the highly reversible H2-activation and permits concrete classification for the first time. The influence of the acid strength on the metal-free hydrogenation of selected olefins is investigated by kinetic experiments and quantum chemical calculations. Detailed information for the rate-determining steps fully support our mechanistic model of a protonation step prior to hydride transfer. The rate of hydrogenation is strongly dependent on the electronic nature of the phosphine and of the acidity of the corresponding phosphonium cation. A careful balance of these two factors provides highly efficient metal-free hydrogenation catalysts. The provided findings are used to revise the reactivity of Lewis bases in the hydrogenation of imines, one of the most recognized application of FLPs.
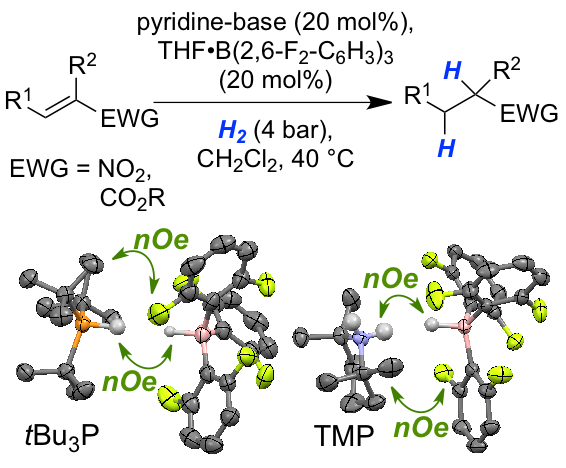
Weak Lewis acid for high nucleophilicity: The B(2,6-F2-C6H3)3-derived hydridoborate, formed from FLP-mediated H2-activation, displayed significant hydridic character. Solid-state and solution structure analysis revealed a dihydrogen-bonded aggregate. The new FLP-system was applied as catalyst in the hydrogenation of nitroolefins and acrylates. The reduced Lewis acidity provides higher reactivity and functional group tolerance in FLP-chemistry.
SYNPACTS: The frustrated Lewis pair (FLP) catalyzed hydrogenation of organic molecules is discussed. The saturation of polarized double bonds by FLP can be described as the nucleophilic addition of hydrides to the polar double bond prior to proton transfer. In contrast, the hydrogenation of olefins proceeds first by protonation forming a transient carbocation, which is subsequently attacked by the hydride. Both processes give rise for efficient conversion of unsaturated organic compounds by a metal-free methodology employing molecular hydrogen.
Dieser Artikel wurde zwei Mal in Chemical & Engineering News und zwei Mal in Nachrichten aus der Chemie herausgehoben:
• "Metal-Free Olefin Hydrogenations" C&EN, 2012, 90, 27.
• "Frustrated Lewis Pairs Go Hydrogenating" C&EN, 2012, 90, Issue 52, 21.
• "Kalt und Gut" Nachr. Chem. 2013, 61, 107.
• "Trendberichte 2012: Organische Chemie " Nachr. Chem. 2013, 61, 293.
Weak Nucleophiles for Strong Activation: The reversible activation of dihydrogen by an electron deficient phosphine, (C6F5)PPh2 (2), in combination with the Lewis acid B(C6F5)3 (1) at –80 °C was accomplished. Such systems exhibit novel reactivity so that the catalytic hydrogenations of double bonds were achieved according to a protonation followed by hydride attack pathway. Electron deficient phosphines and diarlyamines were demonstrated to be viable Lewis bases for the hydrogenation of double bonds allowing catalyst loadings of 10 to 5 mol%.
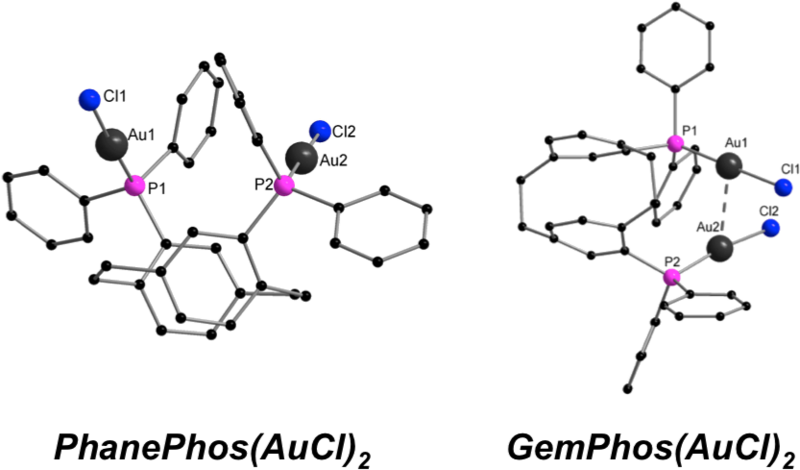
[PhanePhos(AuCl)2] and [GemPhos(AuCl)2] show a very similar ligand scaffold, but different (aurophilic vs. non-aurophilic) intramolecular Au-Au distances. Absorption and PL spectra of both compounds are quite similar. Theoretical investigations reveal that the excited states are of a different character (i.e., influenced by the Au-Au contacts). [PhanePhos(AuCl)2] and [GemPhos(AuCl)2] were also used as catalysts in the intra- and intermolecular hydroamination of alkynes. Good to quantitative conversions on a reasonable time scale were observed. The overall performance of these catalysts in the studied reactions was similar showing that the Au-Au contacts do not have a major influence on the catalytic performance.
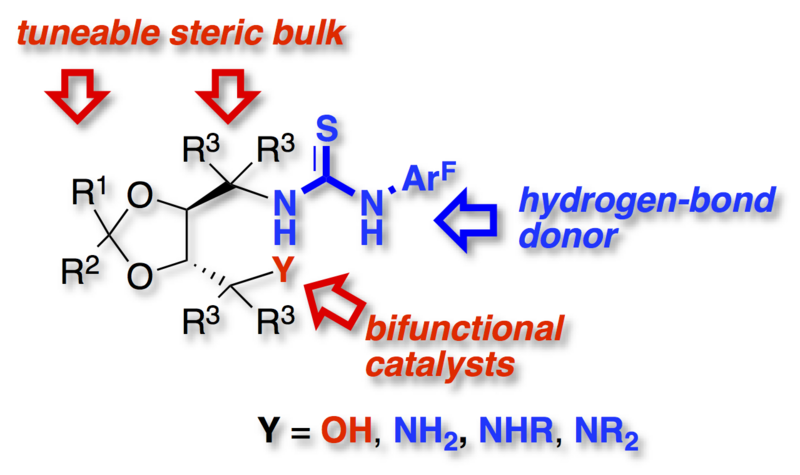
A flexible synthesis of bifunctional thiourea derivatives based on the TADDOL framework is described. Hydroxy as well as primary, secondary and tertiary amino substituted bifunctional hydrogen-bond donors were synthesized.
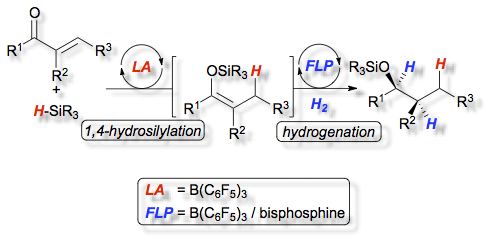
The heterolytic splitting of hydrogen by two types of [2.2]paracyclophane derived bisphosphines (1, 2a and 2b) in combination with tris(pentafluorophenyl)borane (3) at room temperature is described. The corresponding frustrated Lewis pairs (FLPs) exhibit different behavior in the activation of hydrogen. This results from diverse steric and electronic properties of the bisphosphines. The reactivity of the frustrated Lewis pairs was exploited in the first diastereoselective domino hydrosilylation/hydrogenation reaction catalyzed by FLPs.
[14] "Development of [2.2]Paracyclophane derived Hydrogen-Bond Receptors" J. F. Schneider, J. Paradies* Isr. J. Chem. 2012, 52, 76-91.
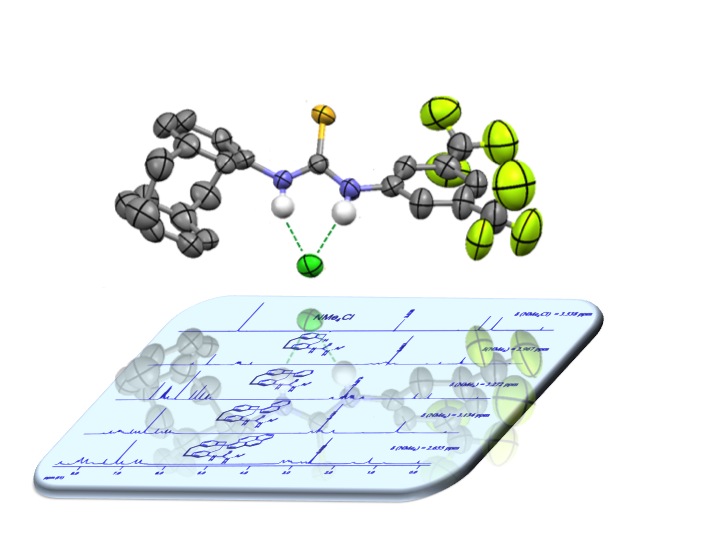
The development of planar-chiral hydrogen-bond donors based on the [2.2]paracyclophane scaffold is discussed. General strategies to access functionalized enantiopure [2.2]¬para-cyclophane derivatives are briefly reviewed, with the focus on suitable precursors for the synthesis of planar-chiral thiourea derivatives. The synthesis of fourteen hydrogen-bond donors is described. The interaction of four thiourea derivatives with hydrogen-bond acceptors (DMSO and tetramethyl-ammonium chloride) was investigated by 1H NMR spectroscopy and X-ray crystallography. A selection of enantio-merically pure planar-chiral derivatives was applied in asymmetric hydrogen-bond catalysis.
[13] "[2.2]Paracyclophane derivatives: Synthesis and Application in Catalysis" J. Paradies* Synthesis 2011, 3749-3766.
Advances in the field of [2.2]paracyclophane chemistry are reviewed including syntheses, chiral resolution and application in organic synthesis. Transition-metal catalyzed as well as organocatalytic transformations are presented focus¬ing on the development of [2.2]paracyclophane derived ligands and catalysts.
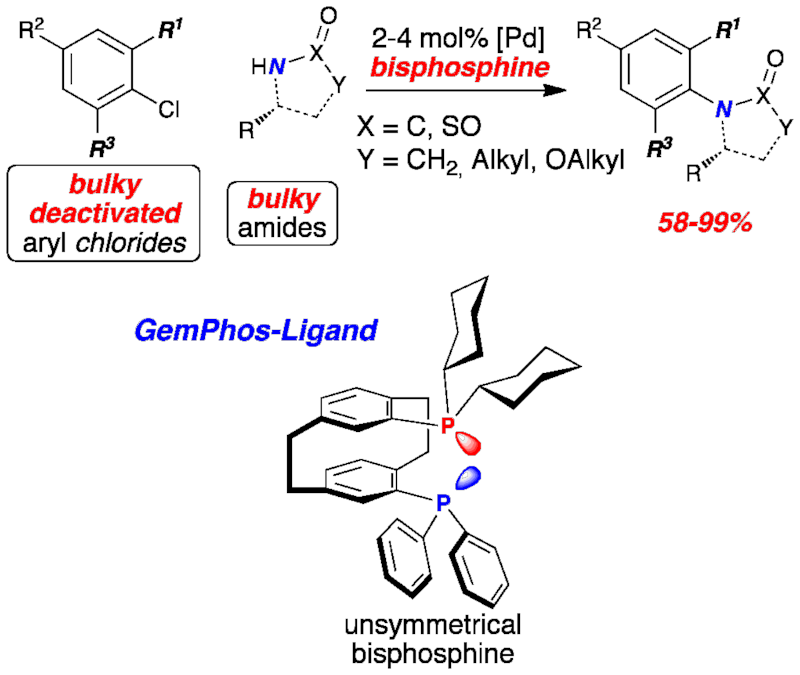
Voluminous amides were coupled with deactivated, sterically hindered aryl chlorides in excellent yields providing products, which have not been efficiently accessible by transition metal catalysis so far. Application of an unsymmetric bisphosphine ligand was critical for the high catalyst activity.
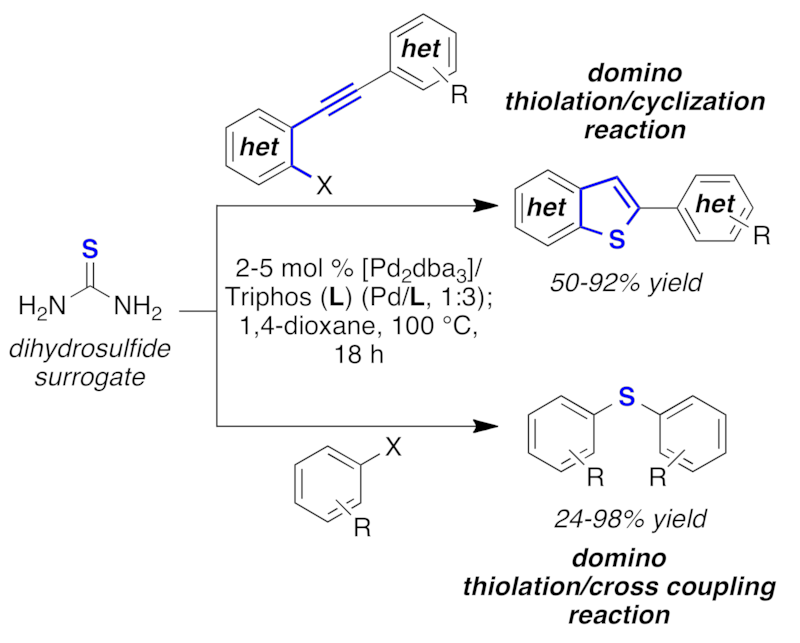
The first C-S bond formation/cross-coupling/cyclization domino reaction using thiourea as a cheap and easy to handle dihydrosulfide surrogate has been developed. Structurally important biarylthioether, benzo[b]thiophenes, and thieno[3,2-b]thiophene scaffolds are provided in high yield.
[10] "Readily Available Hydrogen-Bond Catalysts for the Asymmetric Transfer Hydrogenation of Nitroolefins" J. F. Schneider, M. B. Lauber, V. Muhr, D. Kratzer, J. Paradies* Org. Biomol. Chem. 2011, 9, 4323-4327.
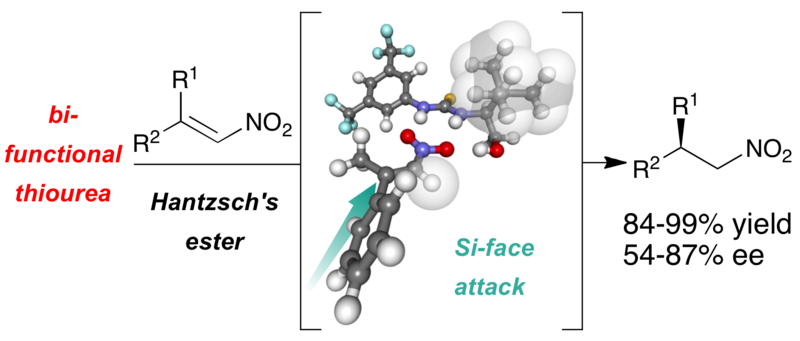
This paper focuses on readily accessible thiourea hydrogen bond catalysts derived from amino acids, whose steric and electronic features are modulated by their degree of substitution at the carbinol carbon center. These catalysts were applied in the asymmetric transfer hydrogenation of nitroolefins furnishing the chiral products in up to 99% yield and 86% enantiomeric excess. The proposed catalyst’s mode of action is supported by mechanistic investigations.
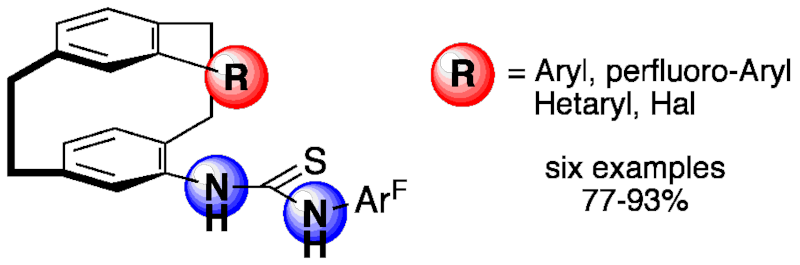
An efficient access to enantiopure pseudo-geminally substituted 4-amino-13-bromo[2.2]paracyclophane is described. The aminobromide was employed in cross-coupling reactions to yield arylated 4-amino[2.2]paracyclophanes. The amines were converted into enantiopure planar-chiral thioureas, which are potential hydrogen-bond donors for enantioselective organocatalysis.
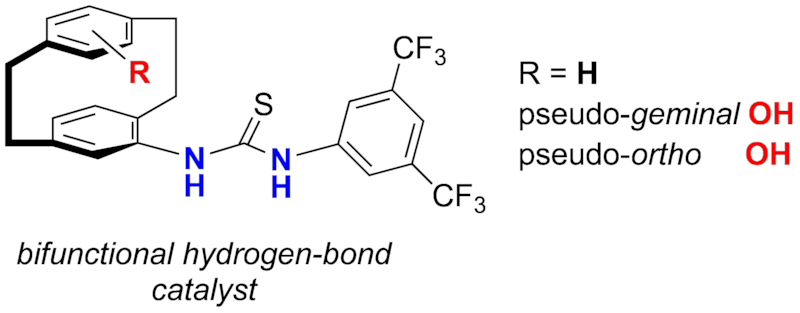
The first planar-chiral thiourea hydrogen-bond catalysts were synthesized from the corresponding [2.2]paracyclophanylamines. Enantiopure bifunctional thioureas operate as hydrogen-bond catalysts and display enhanced activity in comparison to their monofunctionalized derivatives.
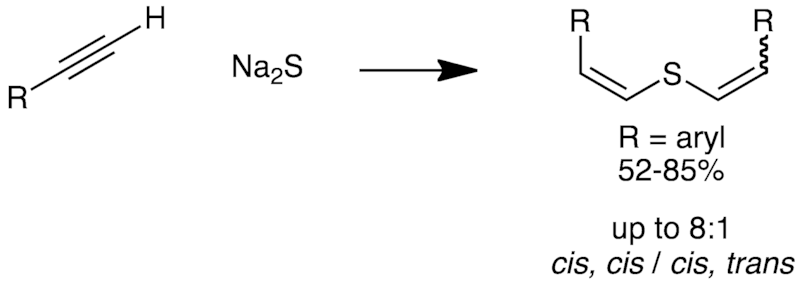
[7] "Synthesis of Divinylsulfides" J. Paradies* Synthesis 2010, 947-942.
Arylacetylenes react with sodium sulfide in the presence of water to yield divinylsulfides. The reaction proceeds in good to excellent yield for electron-neutral and electron-deficient aromatic systems. Three selected divinylsulfide derivatives were oxidized selectively to the corresponding sulfoxides or sulfones.
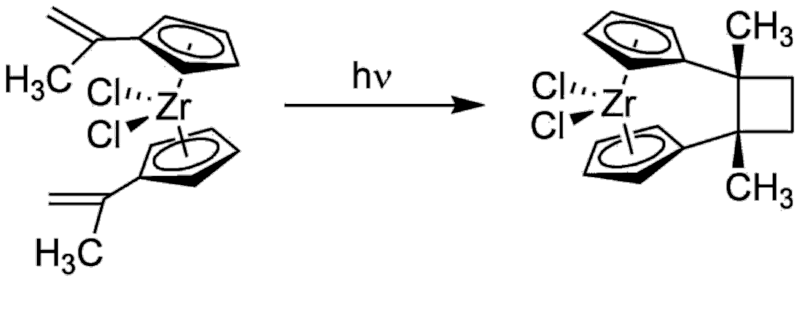
Photolysis of Bis(1-methylethenyl-cyclopentadienyl)-zirconium dichloride at 0 °C with Pyrexfiltered UV light resulted in a rapid and complete intramolecular [2+2]cycloaddition reaction to yield the corresponding cyclobutylene- bridged ansa-zirconocene dichloride isomer.
[5] "Developing a Functional Group Chemistry of Organolithium Compounds: [2+2] Cycloaddition of Alkenyl-substituted Li-Cyclopentadienides" J. Paradies, G. Erker,* R. Fröhlich, Angew. Chem. 2006, 116, 3150-3153; Angew. Chem. Int. Ed. 2006, 45, 3079-3082.
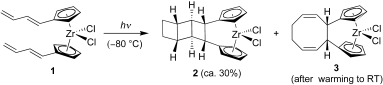
Coming together at the ends:
Bis(butadienyl-cyclopentadienyl) dichlorozirconium (1) undergoes dynamic topochemical reactions upon photolysis that eventually lead to the formation of ladderane (2) derivative and the organometallic cyclooctadiene derivative (3).
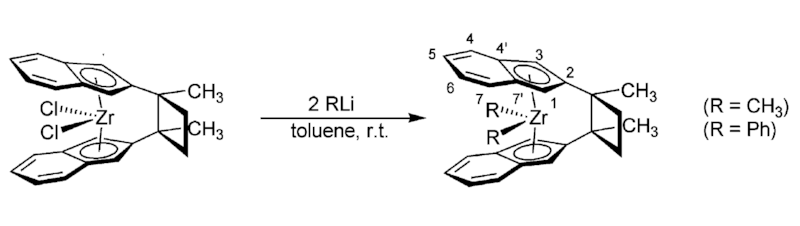
[4] “Insertion Reactions at Cyclobutylene-Bridged ansa-Metallocene Complexes: A Quest for the Influence of Covering Phenylene Units” L. Chen, W.-L. Nie, J. Paradies, G. Kehr, R. Fröhlich, K. Wedeking, G. Erker* Organometallics, 2006, 25, 5333-5344.
The cyclobutylene-bis(2-indenyl)zirconium dichloride complex, derived from an intramolecular photochemical [2+2] cycloaddition reaction of bis[2-(methylethenyl)indenyl]zirconium dichloride, was reacted with methyllithium or phenyllithium to yield the corresponding cyclobutylene-bis(2-indenyl)- zirconium dimethyl or diphenyl complex, respectively.

Photolysis of bis[1,3-di(1-methylethenyl)C5H3]zirconium dichloride with Pyrex-filtered UV/vis light at ambient temperature led to a rapid intramolecular [2+2]- cycloaddition reaction to yield a 1:1 mixture of the meso- and rac-isomers of the corresponding singly cyclo-butylene-bridged ansa-metallocene system. Photolysis of this mixture with quartz-filtered UV light at -80 °C very slowly converted meso-4a to rac-4a. Over 2 days a 12:1 ratio of rac/meso-4a was achieved under these conditions.
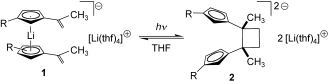
[2] “Developing a Functional Group Chemistry of Organolithium Compounds: [2+2] Cycloaddition of Alkenyl-substituted Li-Cyclopentadienides” J. Paradies, G. Erker,* R. Fröhlich Angew. Chem. 2006, 116, 3150-3153; Angew. Chem., Int. Ed. Engl. 2006, 45, 3079-3082.
Li and light:
Alkenyl-substituted lithium cyclopentadienides, which are in equilibrium with the substituted lithocene anion structure 1, undergo a photochemical [2+2] cycloaddition to yield selectively the carbon-carbon coupling product 2. This is a rare case of organic functional-group chemistry for a reactive organolithium compound.
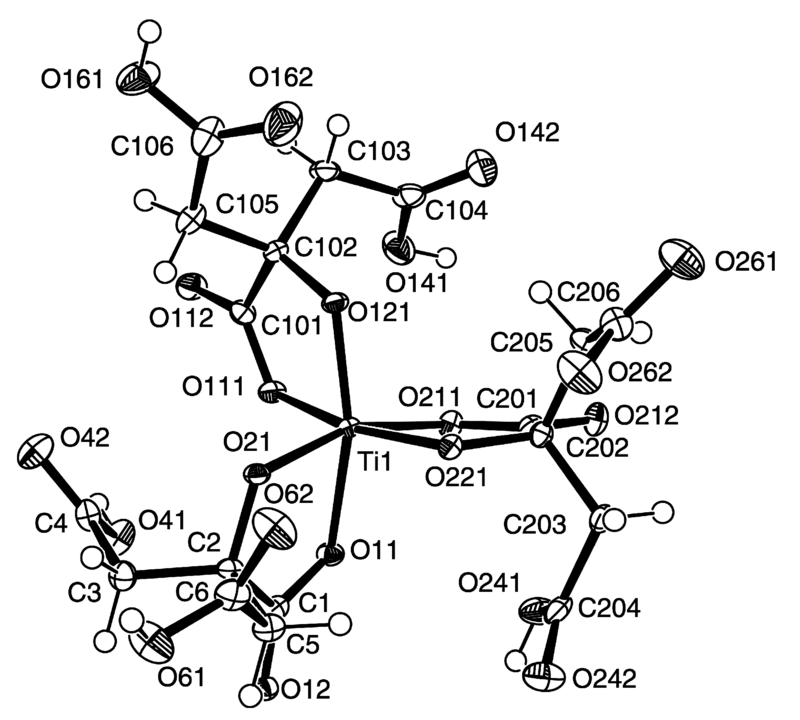
We show that Ti(III) citrate is generated in a facile manner and in good yield by the action of UV radiation on Ti(IV) citrate in aqueous solution. The Ti(III)-citrate species formed was isolated and characterised by UV–Visible spectroscopy, showing an absorption at 547 nm (e = 100 M1 cm1), and by electron paramagnetic resonance (EPR) spectroscopy giving a resonance at g = 1.949 (linewidth = 60 G).
