
Research Concept
Chemical as well as physical processes are intrinsically associated with large length and time scales. Thus, an at least partially quantum mechanical description of such a many-body system is analytically only possible in very few exceptional cases. Instead, a statistical mechanical treatment with quantum mechanical methods that can be solved by modern massively parallel high-performance computers is required. The main task is therefore to devise and implement novel numerical techniques, which are as efficient as possible and yet, at the same time, qualitatively reproduce the correct chemistry and physics of the original system.
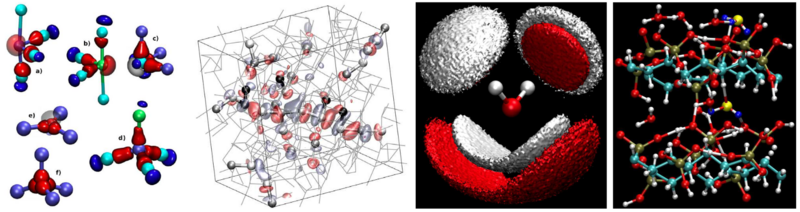
However, our purpose is not solely the development of new algorithms, but to solve scientifically relevant questions of chemistry, physics, material sciences and biophysics. In general our main interest is the investigation of complex systems in condensed phases (liquids, solids and supramolecular systems). In particular, our research group focuses on studying aqueous systems such as water interfaces, water in confined geometries, biological relevant reactions in aqueous solution and the heterogenous “on-water” catalysis. Additionally, we are also investigating sustainable systems and energy materials, specifically CIGS-based thin-film solar cells, polymer electrolyte fuel cells, lithium-sulfur batteries, novel hydrogen-storage materials, solid hydrogen, non-volatile phase-change materials and topological Weyl-semimetal-based catalysis.
Method development
Our methodological focus is on the development and application of the molecular dynamics technique at finite temperatures on large length and time scales, where the atomic forces are calculated "on-the-fly" by parameter-free electronic structure methods. In particular, the second generation Car-Parrinello ab-initio molecular dynamics method, which we developed, allowed to study phenomena that previously were thought to be not feasible. Furthermore, we have also focused on so-called linear scaling electronic structure approaches for metallic systems. In contrast to conventional tight-binding or density functional theory based electronic structure methods, this development permits to study very complex systems with up to a million atoms quantum-mechanical. In addition to combining this technique with the already alluded to second generation Car-Parrinello ab-initio molecular dynamics method, our main emphasis was the development and application of a novel energy decomposition analysis scheme for periodic systems. With this new scheme we are now able to study the various interactions between the atoms, which are involved in chemical bonding, in great detail. Moreover, we are working on inverse design methods, which permits topredict in silico novel energy materials with predetermined properties from first-principles.
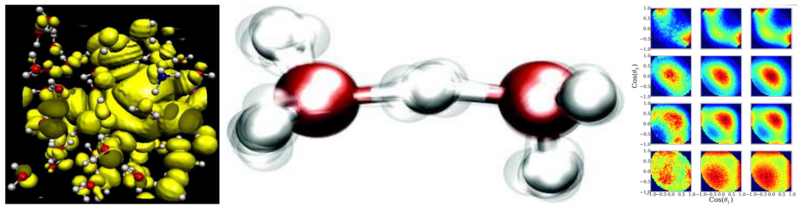
Currently, the main topic is the development of novel theoretical techniques to accelerate the so-called ring-polymer path-integral molecular dynamics technique in order to compute the quantum dynamics of complex systems. At variance to already existing path-integral simulations this scheme not only allows for an exact treatment of nuclear quantum effects, which are especially important for lightweight atoms, but also yields the correct real time evolution of the atoms. By applying the so-called “ring contraction method”, it is nowadays possible to simulate exceedingly complex systems using classical force fields while taking the harmonic and anharmonic zero point energy, as well as quantum mechanical tunneling effects explicitly in to account at minimal additional computational cost. Very recently, we succeeded in transferring this idea to the second generation Car-Parrinello ab-initio molecular dynamics method and to combine it with parameter-free “on-the-fly” electronic structure calculations, which enables us to conduct full quantum dynamics simulations and in particular to simulate vibrational spectroscopies such as IR, Raman and SFG spectroscopy at de facto no additional computational cost in comparison to ab-initio molecular dynamics.
Applications
For us, the application of the simulation methods we develop to scientifically relevant questions of chemistry, physics, material sciences and biophysics is of uppermost importance. On the one hand as the definite demonstration of their utility in rationalizing and predicting new phenomena, but on the other hand also to inspire new methodological developments. In general, our current and future research projects are concerned with complex systems in condensed phases. In particular with liquid, liquid/solid, as well as solid state systems. While in the latter case covalent bonds specify the structure of a system and its properties, in the former they are determined by weak intra- and intermolecular interactions.
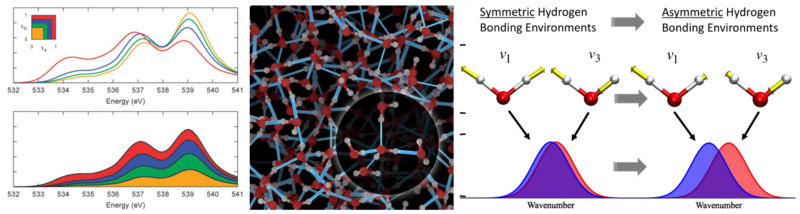
In the case of liquids we are in our research group particularly interested in aqueous systems. Its corresponding hydrogen network undergoes ultrafast fluctuations on the femtosecond timescale and is decisive for the internal stabilization of biomolecules and catalysis of reactions. These fluctuations give rise to a characteristic spectroscopic fingerprint, which is directly accessible by experiment. We therefore aim at understanding the relationships between the dynamics and structure of the hydrogen bond network and spectroscopy. For instance, combining our recently developed energy decomposition analysis method with second generation Car-Parrinello ab-initio molecular dynamics and XAS calculations, we have recently been able to settle the longstanding controversy, whether liquid water is four- or rather two-fold coordinated. Furthermore, we have established a quantitative relationship between the anisotropy of the proton magnetic shielding tensor and the covalency of hydrogen bonding, which facilitates to experimentally determine the strength of hydrogen bonding by means of NMR measurements. For the purpose to elucidate experimental vibrational spectra we have also devised a new analysis method. Based on that, we are increasingly studying liquid water in rather complex geometries, such as the metal oxide/water interface and the heterogenous “on-water” catalysis.
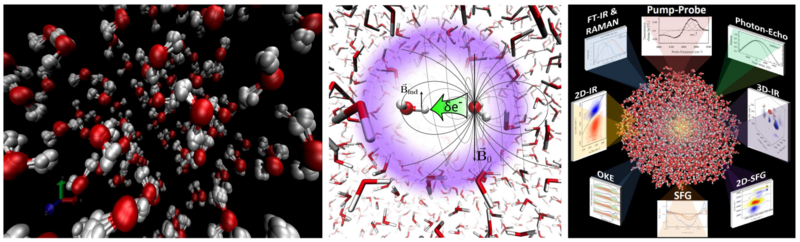
Moreover, we give our attention to study crystalline and amorphous solid state systems, such as hydrogen-rich systems and disordered glasses, which moreover can also be applied to determine the structure of folded proteins. With respect to amorphous glasses, we devote ourself to determine the structure of so-called phase change materials, which due to their high optical, electrical and magnetic contrast are ideally suited as future non-volatile memory devises. Based on second generation Car-Parrinello ab-initio molecular dynamics simulations we have been able to successfully predict the atomic structures for a variety of rather complex amorphous glasses and explain the underlying mechanism of phase-change materials. Due to the fact that this approach is computationally still rather expensive, we therefore plan to directly predict novel materials with predetermined target properties using our recently developed “inverse design” methods in conjunction with accurate electronic structure calculations and experimental measurements